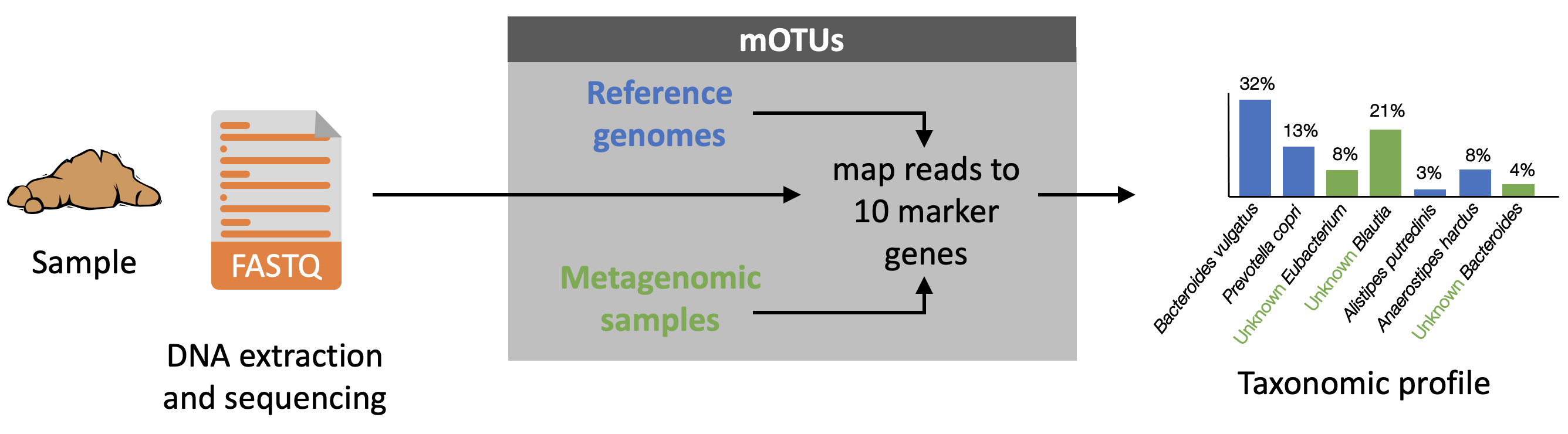
microbiomics tools
Official website: https://motu-tool.org/
Github: https://github.com/motu-tool/mOTUs
Papers:- mOTUs1: Sunagawa et al. Nature Methods 2013
- mOTUs2: Milanese, Mende et al. Nature Comm 2019
- protocol: Ruscheweyh et al. Current protocols 2021
App website: https://microbiomics.io/tools/mbarq-app/
Github: https://github.com/MicrobiologyETHZ/mbarq
Paper: Sintsova, Anna et al. bioRxiv 2023
Description: DNA barcoding has become a powerful tool for assessing the fitness of strains in a variety of studies, including random transposon mutagenesis screens, attenuation of site-directed mutants, and population dynamics of isogenic strain pools. However, the statistical analysis, visualization and contextualization of the data resulting from such experiments can be complex and require bioinformatic skills. Here, we developed mBARq, a user-friendly tool designed to simplify these steps for diverse experimental setups. The tool is seamlessly integrated with an intuitive web app for interactive data exploration via the STRING and KEGG databases to accelerate scientific discovery. Graphical abstract: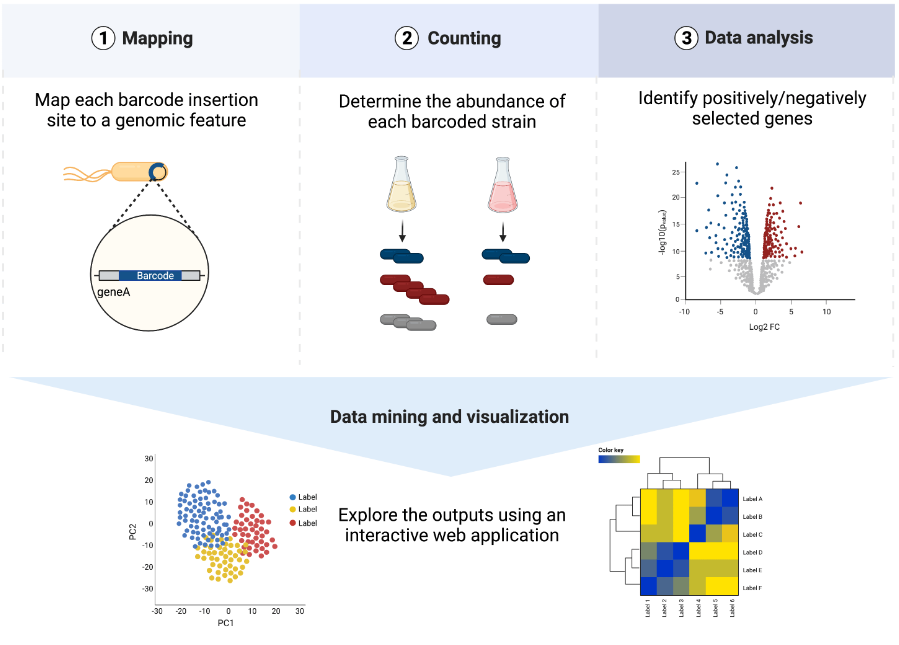
Github: https://github.com/SushiLab/mTAGs
Paper: Salazar, Ruscheweyh et al. Bioinformatics 2021
Description: mTAGs is a tool for the taxonomic profiling of metagenomes. It detects sequencing reads belonging to the small subunit of the ribosomal RNA (SSU-rRNA) gene and annotates them through the alignment to full-length degenerate consensus SSU-rRNA reference sequences. The tool is capable of processing single-end and pair-end metagenomic reads, takes advantage of the information contained in any region of the SSU-rRNA gene and provides relative abundance profiles at multiple taxonomic ranks, including OTUs defined at a 97% sequence identity cutoff. Although the primary use of mTAGs is the taxonomic profiling of metagenomes, it can also be used for profiling SSU-rRNA amplicon data or for classifying amplicon sequence variants. Graphical abstract: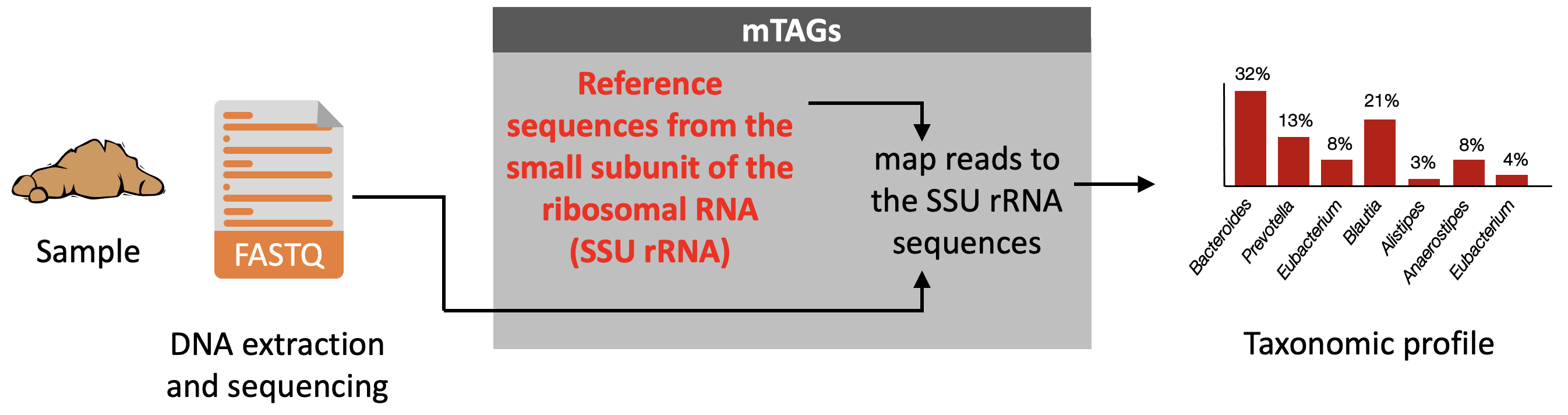

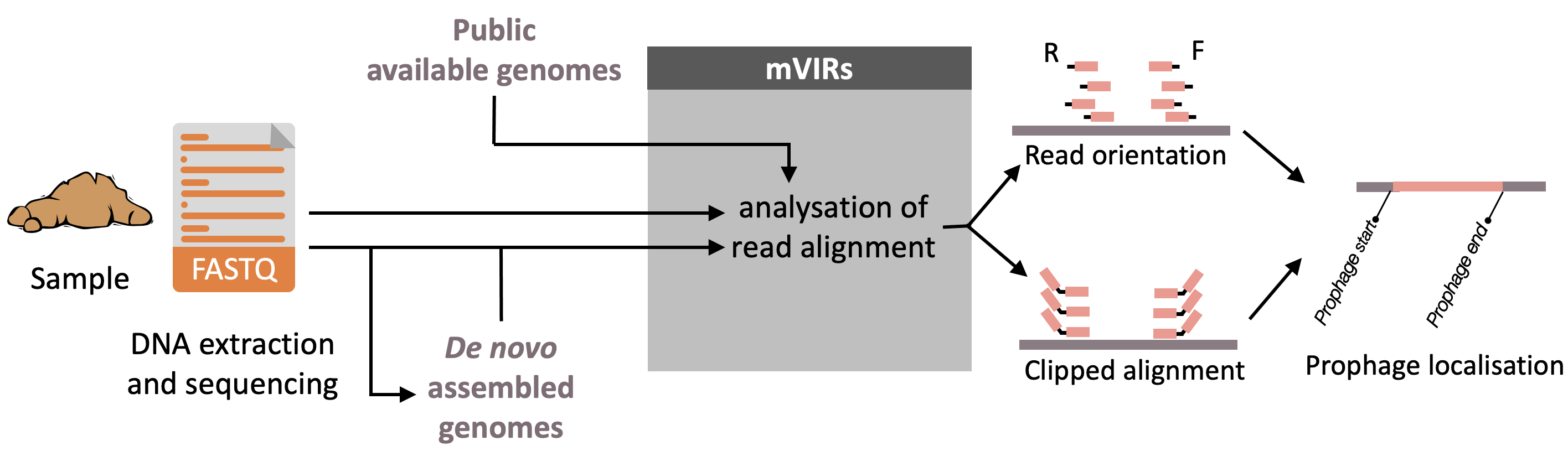
Github: https://github.com/SushiLab/mVIRs
Paper: Zünd et al. Microbiome 2021
Description: VIRs is a bioinformatics tool to locate the integration sites of inducible mobile genetic elements, mainly prophages, in microbial genomes. It exclusively identifies prophages found in circularised or concatena ted form after the induction. The prophages are detected by analysing the orientation and apparent insert size of paired-end reads aligned to a reference genome and locates the exact genomic coordinates by partially aligned reads. mVIRs a pplies to microbial community samples, using either publicly available or de novo assembled genomes as references for the alignment of sequencing reads. The output of mVIRs can be quality checked and annotated by tools, such as checkV or VirSorter2 and PHANOTATE, to identify putative prophage candidates and builds the foundation to quantify phage-to-host ratio as relative prophage activity levels. Graphical abstract: